Abstract
Lactic acidosis is a serious and frequently encountered condition in oncological critical care. Although it is commonly associated with generalized or localized tissue hypoperfusion, metabolic and drug-related causes should also be considered. Early recognition and appropriate management are essential for improving outcomes in critically ill patients. We report a case of refractory lactic acidosis associated with unexplained hypoglycemia, hypertriglyceridemia, and hyperuricemia. Common causes of type A lactic acidosis, including hypoperfusion, anemia, hypoxia, and seizures, were excluded. Other potential causes of type B and D lactic acidosis, such as liver disease and medication effects, were also ruled out. The patient had no known history of metabolic disease during childhood; however, the clinical presentation and laboratory findings were highly suggestive of glycogen storage disease type Ib (GSD-Ib).
GSD-Ib is a rare autosomal recessive metabolic disorder caused by mutations in the glucose-6-phosphate translocase (G6PT) gene located on chromosome 11q23. The resulting defect impairs both glycogenolysis and gluconeogenesis, leading to metabolic abnormalities including hypoglycemia, hyperlactatemia, hyperuricemia, and hypertriglyceridemia. GSD-Ib Patients commonly develop neutropenia, which predisposes them to recurrent infections and often requires treatment with granulocyte colony-stimulating factor (G-CSF). Long-term G-CSF therapy has been associated with an increased risk of myeloid malignancies, particularly acute myeloid leukemia (AML).
Glycogen storage diseases are genetic disorders that are typically diagnosed in childhood, making diagnosis in adulthood extremely rare. In this case, a late genetic mutation was suspected to have resulted in adult-onset GSD-Ib and malignancy. This rare presentation suggests a possible link between GSD-Ib and AML. Therefore, surveillance for glycogen storage disease in patients with AML, and vice versa, may be warranted. This association should be considered in critically ill AML patients presenting with refractory lactic acidosis after exclusion of other etiologies.
Keywords: AML, Acute myeloid leukemia, Lactic acidosis, Hypoglycemia, Cancer, Glycogen storage disease, Hypertriglyceridemia
Introduction
Lactic acidosis is a significant concern in oncological critical care, as it can result from generalized or local hypo perfusion, or other underlying causes. Prompt identification and management of lactic acidosis are crucial for improving outcomes in critically ill patients. We present a case of refractory lactic acidosis associated with unexplained hypoglycemia, hypertriglyceridemia, and hyperuricemia. After excluding hypo perfusion, anemia, hypoxia, seizures, or any other causes of Type A lactic acidosis, as well as liver diseases, medications, or other factors contributing to Type B or D lactic acidosis (1), we considered that the patient’s clinical presentation and investigations best align with adult-onset Glycogen Storage Disease Type Ib.
Glycogen Storage Disease Type Ib (GSD-Ib) is one of over 12 inherited metabolic disorders that affect glycogen metabolism. It is an autosomal recessive condition caused by mutations in the glucose-6-phosphate translocase (G6PT) gene located on chromosome 11q23 (2). G6PT deficiency disrupts both glycogenolysis and gluconeogenesis. Patients with GSD-Ib typically experience hepatomegaly, growth retardation, hypoglycemia, hyperlactatemia, hyperuricemia, and hyperlipidemia in early infancy. In addition, they develop neutropenia, which increases their risk of severe infections (3).
To manage these complications, long-term treatment with granulocyte colony-stimulating factor (G-CSF) is often used. However, this treatment can increase the risk of acute myeloid leukemia (AML) or myelodysplastic syndromes (MDS) in patients with inherited bone marrow failure conditions such as severe congenital neutropenia (4). The development of these myeloid malignancies is often linked to cytogenetic abnormalities involving chromosome 7, and G-CSF is known to promote the proliferation of monosomy 7 cells in vitro.
Although glycogen storage diseases are typically diagnosed in childhood due to their genetic basis, our patient was diagnosed in adulthood with a mutation that led to Glycogen Storage Disease Type Ib and malignancy. There have been several reports in recent years of patients with GSD-Ib developing AML after prolonged treatment with G-CSF. However, it is rare for this association to be observed in adult patients with leukemia and adult-onset GSD-Ib.
Given the potential strong association, surveillance for both Glycogen Storage Disease and acute myeloid leukemia in such patients is essential. This association should be considered in cases of refractory lactic acidosis in critically ill patients with acute myeloid leukemia, particularly after excluding other etiologies of lactic acidosis.
Case Presentation
A 52 years old male patient who is not known with any cardiovascular, endocrinal, renal, hepatic disorders, familial hyperlipidemia, congenital anomalies or abnormal milestones, noticed a lump over his chest wall, superficial ultrasound was done and revealed a hypoechoic mass with surrounding edema.
A biopsy was taken revealed fibrous tissue infiltrated by a tumor formed of small round cells with extensive crushing possessing hyperchromatic nuclei. The immature cells are positive for CD43, KCA, TdT and negative for MPO, CD117, CD 56, CD 20, CD30 and CD3 by immunohistochemical stains confirming a diagnosis of myeloid sarcoma.
Initial bone marrow biopsy revealed hypercellularity with dysplastic features and a blast count of 20%, which was confirmed by flow cytometry with 30% CD34-positive cells. A second bone marrow biopsy showed 21% blasts and persistent dysplastic features. Cytogenetic analysis revealed monosomy 7 in 33 of 34 metaphases and a translocation t(3;8)(q26;q24).
A diagnosis with chest wall myeloid sarcoma which is a subtybe of Acute Myeloid Leukemia is then confirmed. The patient received 3+7 protocol of chemotherapy “Daunorubicin and Cytarabine”. After 3 months the patient came to the ER department at Egyptian National Cancer Institute and was presenting with persistent vomiting and fatigue.
Thorough medical & drug history taking and full body examination were done revealing a history of persistent vomiting for the last month, abnormal lipid profile with extremely high serum levels of triglycerides exceeding 3000 mg/dl. This lab test was done at the last day of Ramadan 2024 “a month for MUSLIMS intended fasting to cultivate self-discipline, spiritual growth, and empathy for the less fortunate” which preceded the patient admission at our institute critical care unit by 1 month. It`s worthy to be mentioned, that the patient even didn`t receive any kind of treatment for hypertriglyceridemia.
The patient was admitted our critical care unit. He was fully conscious, oriented to time, place and persons. His calf muscles were lax, intact peripheral pulsations, clear chest by auscultation and no abdominal pain or tenderness. His hemodynamics on admission were as follow; HR 90 beats/min, RR 20 breaths/min, BP 120/80 mmHg, Temp 37°C, RBS 70 mg/dL.
Full labs were done on admission; CBC showed pancytopenia: WBC 1900/μl, hemoglobin 6.7 g/dl, and platelet count 86 × 103/μl, with a differential count of 61% lymphocytes, 5% monocytes, 4% mature neutrophilic granulocytes, and 30% blasts.
Also hyperlactatemia (19mmol/l), high anion gap metabolic acidosis with anion gap 26, hyperuricemia (11mg/dl), hypertriglyceridemia (850mg/dl), while the renal function tests, liver function tests, sepsis markers, and electrolytes were average apart from mild hypokalemia (Serum potassium = 3.3mmol/L).
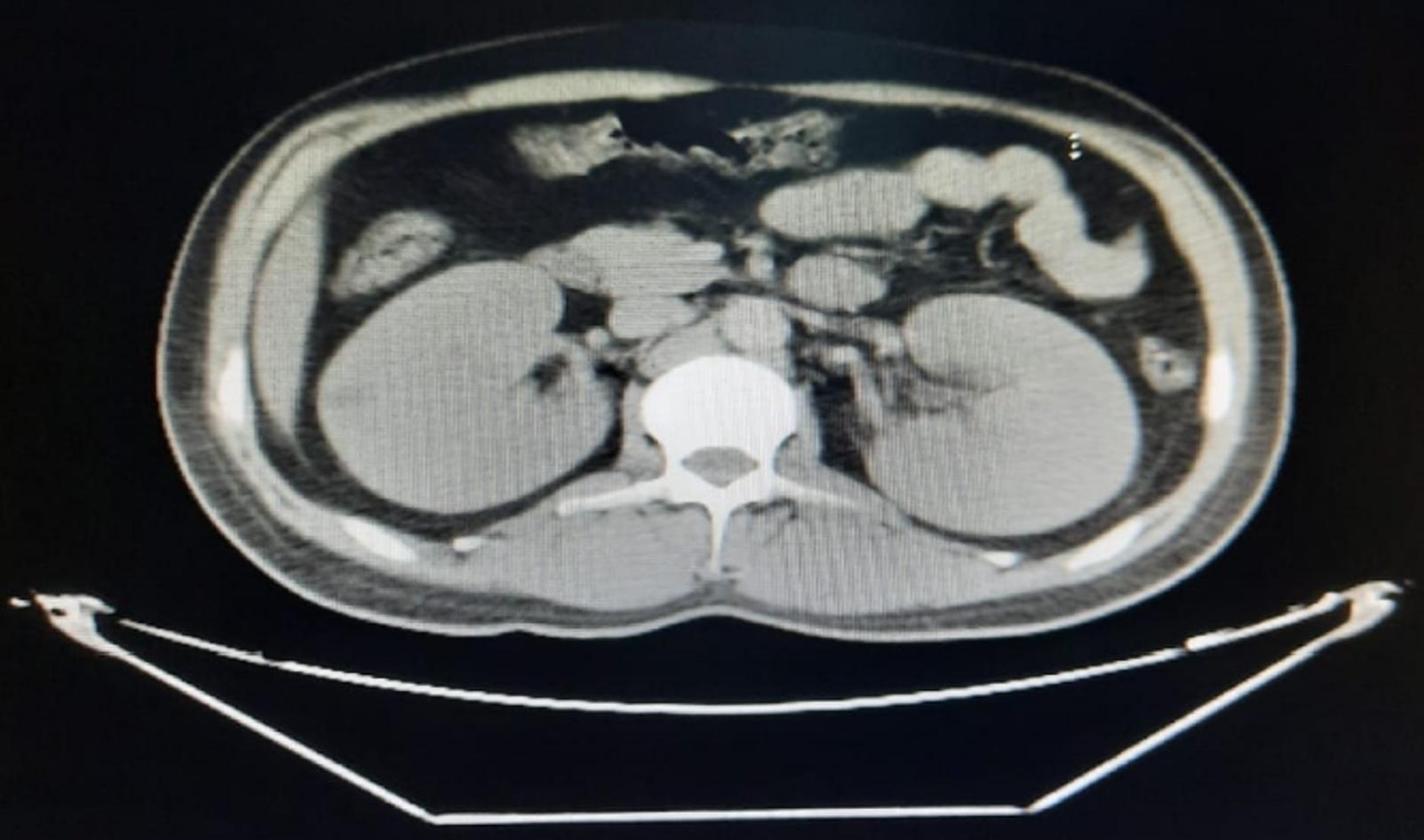
CT Abdomen with contrast was done and ruled out pancreatitis, but bilateral almost symmetrical nephromegaly were very evident (Figure 1).
The patient received crystalloids and insulin infusion to lower serum triglycerides’ level as insulin enhances lipoprotein lipase which is the crucial enzyme for the hydrolysis of triglycerides. Dextrose 25% infusion was added to support serum normoglycemia.
An approach for lactic acidosis was followed and the hyper lactatemia wasn’t neither explained by type A or type B nor type D lactic acidosis causes as the patient was not shocked, or hypoxic, or severely anemic, or taking any medications, or hepatic or suffering from any kind of local hypo perfusion. Also, the patient has no history of any bariatric or intestinal surgeries. Warburg effect of leukemic cells which is rare could not explain hypoglycemia, hyperuricemia and hypertriglyceridemia, leaving a question mark about the etiology of his presentation with severe lactic acidemia without any identifiable reason.
On the 3rd day of ICU stay with continuous crystalloids and insulin/dextrose 25% infusion, the serum lactate level dropped to 12mmol/l, improved metabolic acidosis with anion gap 18, serum triglycerides` serum level declined to 370mg/dl and then, Insulin infusion was stopped and instead, statin and fenofibrate were added.
The patient`s serum glucose level, for couple of days, was inappropriately steady low not exceeding 60mg/dl in spite of withholding insulin infusion, receiving 200cc of dextrose 25% infusion hourly and adding sources of simple carbs to his meals but it didn’t make any difference in his serum glucose levels leaving another question mark about his condition.
Serum C-Peptide level, Thyroid Profile, Cortisol level were normal excluding an insulinoma, hypothyroidism and hypoadrenalism respectively.
Classic Clinical Presentation of Hypertriglyceridemia, Hypoglycemia, Hyperlactatemia, Hyperuricemia, Nephromegaly and Neutropenia raised a suspicion of Glycogen Storage Disease Type Ib. Due to financial issues, we could not confirm diagnosis with genetic sequencing.
Steroids were not added to his treatment plan due to their known effect on uric acid and triglycerides serum levels. The patient was encouraged to avoid fasting at all being the precipitating factor in worsening condition in such a disease. Uncooked starch and other starchy food stuff were added to his nutrition plan, then improvement of serum glucose levels were noted.
Dextrose 25% infusion was discontinued and the patient was discharged to home with slight metabolic acidosis with Anion Gap 14, Lactate 8mmol/l, RBS 70mg/dl, Uric acid 7mg/dl, Triglycerides 250mg/dl.
The patient was then informed with the special nutrition instructions specific for Glycogen Storage Disease Type I and keeping on statins, allopurinol and omega 3, however the patient wasn’t complaint.
1 month later the patient came to the ER department presenting with Severe Dyspnea, Stridor, Drowsiness, Severe Hypoglycemia (20mg/dl), Severe Anion Gap Metabolic acidosis with Anion Gap 30, Profound Neutropenia, Thrombocytopenia and Sepsis.
The patient was resuscitated with boluses of dextrose 25% solution, intubated and mechanically ventilated, Urgent CT neck was done showing vocal cord mass thought to be a vocal cord chloroma (myeloid sarcoma of the larynx) as an extramedullary manifestation of leukemia spreading to soft tissues.
The patient condition was worsened due to development of a septic shock, granulocyte-colony stimulating factor & empirical antibiotics and antifungal were added to combat with sepsis. Neutropenia and dysfunction of neutrophils can be linked to the accumulation of 1,5-anhydroglucitol-6-phosphate (1,5-AG6P). This compound, a potent hexokinase inhibitor, forms slowly in cells from 1,5-anhydroglucitol (1,5-AG), a glucose analog found in the blood. In healthy neutrophils, accumulation of 1,5-AG6P is normally prevented by hydrolysis via G6PC3 after being transported into the endoplasmic reticulum (ER) by G6PT which is deficient in a such condition.
SGLT2 inhibitors, has been reported in previous studies with effectiveness in significant reduction of blood 1,5-AG6P levels, improving neutrophil counts and function.
However, despite the potential benefits of this treatment, it was not included in our patient’s treatment plan due to concerns about hemodynamic instability. Tragically, the patient developed refractory shock and, despite efforts to manage the condition, he did not survive.
Discussion
Glycogen storage disease type I (GSD I), also known as Von Gierke disease, is an inherited disorder caused by deficiencies of specific enzymes in the glycogen metabolism pathway. It comprises 2 major subtypes, GSD Ia and GSD Ib. The incidence of GSD I in the overall population is 1/100,000 with GSD Ia and Ib prevalent in 80% and 20% respectively in a previous study by Nirzar S. Parikh, 2023 (5).
In GSD Ia, there is a deficiency of enzyme glucose-6-phosphatase (G6Pase) which cleaves glycogen to glucose.
In GSD 1b, there is a normal G6Pase enzyme activity but with a deficiency of the transporter enzyme, glucose-6-phosphate translocase (G6PT), GSD Ib results from mutations in the SLC37A4 gene on chromosome 11q23.3.
The lack of either glucose-6-phosphatase (G6Pase) catalytic activity or glucose-6-phosphate transporter SLC37A4 (G6PT) activity in the liver impairs the normal conversion of glucose-6-phosphate to glucose through glycogenolysis and gluconeogenesis (5).
This impairment leads to severe hypoglycemia and a range of symptoms characteristic of Glycogen Storage Disease Type I (GSD I). The deficiency in G6Pase disrupts the final step of glucose production from glycogen, while the deficiency in G6PT prevents the transport of glucose-6-phosphate into the endoplasmic reticulum where it can be converted to glucose (5).
Both deficiencies result in the accumulation of glucose-6-phosphate, which contributes to hypoglycemia and over production of triglycerides, uric acid and lactic acid (5).
High serum triglycerides level may lead to Acute Pancreatitis which is an emergency and could be excluded by a abdomen CT with contrast (6).
Serum triglycerides levels more than 1000mg/dl should be managed by plasmapheresis, Insulin/Dextrose infusion is highly beneficial for serum triglycerides level below 1000mg/dl to boost lipoprotein lipase activity enhancing cellular uptake of triglycerides, while Statins are sufficient for serum triglyceride levels below 500mg/dl. However treating the etiology is a must (7).
Glycogen accumulation in liver and kidneys may lead to hepatic adenomas and nephromegaly up to renal failure.
Fasting is prohibited for those patients, because it activates glycogenolysis and gluconeogenesis pathways for boosting up serum glucose levels, but with G6Pase or G6PT deficiency ,the patient will experience hypoglycemia, hypertriglyceridemia, hyperuricemia, hyperlactatemia and may be lethal high anion gap metabolic acidosis (8).
Although GSD Ia, GSD 1b are similar in their clinical presentations, but they differ in being neutropenia and thrombocytopenia are characteristic findings in GSD Ib.
A recent research by Veiga-da-Cunha et al. (2019) identified that the inability to eliminate 1,5-anhydroglucitol-6-phosphate (1,5-AG6P), a potent inhibitor of hexokinases, which is slowly formed in the cells from 1,5-anhydroglucitol (1,5-AG), a glucose analog that is normally present in blood. Healthy neutrophils prevent the accumulation of 1,5-AG6P due to its hydrolysis by G6PC3 following transport into the ER by G6PT. G6PT deficiency in the endoplasmic reticulum (ER) membrane of neutrophils, leads to a lack of glucose and heightened oxidative stress, impaired respiratory burst, reduced chemotaxis, and disrupted calcium flux contributing to neutropenia and neutrophil dysfunction in GSD Ib. Additionally, these neutrophils are more prone to apoptosis, which exacerbates neutropenia (9).
An understanding of this mechanism has led to a treatment aimed at lowering the concentration of 1,5-AG6P and 1,5-AG in blood by treating patients with empagliflozin, an SGLT2 (sodium-glucose cotransporter 2) inhibitor, which inhibits renal glucose reabsorption. The enhanced urinary excretion of glucose inhibits the 1,5-AG transporter, SGLT5, causing a substantial decrease in the concentration of this polyol in blood, an increase in neutrophil counts and function and a remarkable improvement in neutropenia-associated clinical signs and symptoms (10).
To manage these complications in GSD Type 1b infants, long-term treatment with granulocyte colony-stimulating factor (G-CSF) is frequently used. However, this treatment can elevate the risk of acute myeloid leukemia (AML) or myelodysplastic syndromes (MDS) in patients with inherited bone marrow failures such as severe congenital neutropenia. The onset of these myeloid malignancies is often associated with cytogenetic abnormalities involving chromosome 7, and G-CSF is known to stimulate the proliferation of monosomy 7 cells in vitro.
Cases of GSD-Ib in early infancy who developed acute myeloid leukemia after couple of years of continuous G-CSF treatment have been reported in the last years, but it has rarely been reported among adult patients with leukemia to be associated with an Adult-Onset GSD Ib.
Our patient has lived a healthy childhood and adulthood till he have been diagnosed with Myeloid Sarcoma (A subtype of Acute Myeloid Leukemia) and GSD-Ib 3 months after without taking even one dose of GSF.
Glycogen storage diseases are explained by genetic defects so it is very unlikely to be diagnosed in adulthood, however our patient was thought to had a genetic mutation in his adulthood that led to development of myeloid leukemia and GSD Ib.
Conclusion
Acute myeloid leukemia and GSD-Ib are linked and may complicate each other with or without GSF treatment. As regarding to our observations, we are recommending that both Surveillance of Acute Myeloid Leukemia in patients of GSD-Ib and Surveillance of GSD-Ib in Acute Myeloid Leukemia patients seem to be mandatory.
This association must be kept in mind in a case of refractory lactic acidosis in a critically ill acute myeloid leukemia patient after exclusion of different etiologies of lactic acidosis.
Ethical approval
This study has been approved by the Egyptian National Cancer Institute (approval date: 12.2.2025). Written informed consent was obtained from the participants.
Author contribution
Study conception and design: AS; data collection: SG, AS; analysis and interpretation of results: GA; draft manuscript preparation: AS, SG, GA. The author(s) reviewed the results and approved the final version of the article.
Source of funding
The authors declare the study received no funding.
Conflict of interest
The authors declare that there is no conflict of interest.
References
- Baddam S, Tubben RE. Lactic Acidosis. [Updated 2025 Apr 28]. In: StatPearls. Treasure Island (FL): StatPearls Publishing; 2025. Available at: https://www.ncbi.nlm.nih.gov/books/NBK470202
- Dissanayake VH, Jayasinghe JD, Thilakaratne V, Jayasekara RW. A novel mutation in SLC37A4 gene in a Sri Lankan boy with glycogen storage disease type Ib associated with very early onset neutropenia. J Mol Genet Med. 2011;5:262-3. https://doi.org/10.4172/1747-0862.1000046
- Sim SW, Weinstein DA, Lee YM, Jun HS. Glycogen storage disease type Ib: role of glucose-6-phosphate transporter in cell metabolism and function. FEBS Lett. 2020;594:3-18. https://doi.org/10.1002/1873-3468.13666
- Calip GS, Moran KM, Sweiss KI, et al. Myelodysplastic syndrome and acute myeloid leukemia after receipt of granulocyte colony-stimulating factors in older patients with non-Hodgkin lymphoma. Cancer. 2019;125:1143-54. https://doi.org/10.1002/cncr.31914
- Nana M, Anastasopoulou C, Parikh NS, et al. Glycogen Storage Disease Type I. [Updated 2025 Apr 6]. In: StatPearls. Treasure Island (FL): StatPearls Publishing; 2026. Available at: https://www.ncbi.nlm.nih.gov/books/NBK534196
- Hidalgo NJ, Pando E, Alberti P, et al. Elevated Serum Triglyceride Levels in Acute Pancreatitis: A Parameter to be Measured and Considered Early. World J Surg. 2022;46:1758-67. https://doi.org/10.1007/s00268-022-06533-w
- Poonuru S, Pathak SR, Vats HS, Pathak RD. Rapid reduction of severely elevated serum triglycerides with insulin infusion, gemfibrozil and niacin. Clin Med Res. 2011;9:38-41. https://doi.org/10.3121/cmr.2010.898
- Froissart R, Piraud M, Boudjemline AM, et al. Glucose-6-phosphatase deficiency. Orphanet J Rare Dis. 2011;6:27. https://doi.org/10.1186/1750-1172-6-27
- Veiga-da-Cunha M, Chevalier N, Stephenne X, et al. Failure to eliminate a phosphorylated glucose analog leads to neutropenia in patients with G6PT and G6PC3 deficiency. Proc Natl Acad Sci U S A. 2019;116:1241-50. https://doi.org/10.1073/pnas.1816143116
- Maiorana A, Tagliaferri F, Dionisi-Vici C. Current understanding on pathogenesis and effective treatment of glycogen storage disease type Ib with empagliflozin: new insights coming from diabetes for its potential implications in other metabolic disorders. Front Endocrinol (Lausanne). 2023;14:1145111. https://doi.org/10.3389/fendo.2023.1145111
Copyright and license
Copyright © 2026 The Author(s). This is an open access article distributed under the Creative Commons Attribution License (CC BY), which permits unrestricted use, distribution, and reproduction in any medium or format, provided the original work is properly cited.